
The cholesterol debate through the lens of human genetics
How strong is the genetic evidence supporting LDL as the causal driver of cardiovascular disease?

Lowering low-density lipoprotein cholesterol (LDL-C) has repeatedly proven to successfully reduce cardiovascular risk. Just earlier this year, two clinical trials provided further evidence supporting both more aggressive LDL-C lowering and broader use of stronger pharmacotherapies: Ez-PAVE showed that intensive LDL-C lowering to <55 mg/dl reduced major cardiovascular events in patients with atherosclerotic cardiovascular disease and VESALIUS-CV showed that the PCSK9 inhibitor evolocumab reduced cardiovascular risk even in patients without prior stroke or myocardial infarction.
One might therefore assume that the debate about the clinical value of keeping LDL-C low would now be settled. However, claims of a “cholesterol charade”, discussions around the retracted Keto-CTA study, the film The Cholesterol Code, and a widely circulated case report of an influencer with LDL-C of >500 mg/dl and no evidence of coronary plaque all fuel a persistent social media narrative that LDL-C may not matter, or at least not for everyone.
Beyond clinical trials, human genetic studies provide what is arguably the most compelling evidence that LDL-C, or more precisely apolipoprotein B (ApoB)-containing lipoprotein particles, are the causal drivers of atherosclerosis and cardiovascular risk. This evidence spans multiple independent layers and has actively shaped the currently accepted model of atherosclerosis accumulation. And as is the case with any good model, genetic predictions have been highly successful, directly informing the development of therapeutic strategies.
Why genetics matters
Human genetic studies have key advantages in this context:
(1) They carry a lower risk of confounding than conventional observational studies. Any confounders, such as obesity or smoking, could not have influenced what genetic variants one inherits from their parents.
(2) They influence disease risk in a clear temporal pattern, where the genetic variant precedes the disease by definition, which eliminates the possibility of reverse causation.
(3) Because they are associated with lifelong effects, they can inform on the impact of long-term exposure to high or low LDL-C in a way that clinical trials of short-term interventions cannot.
On the other hand, most genetic variants have very small effects on human phenotypes, and this has been a key source of criticism. In the largest genome-wide association study (GWAS) for LDL-C in 1.65 million individuals, most significant common variants (frequency >1% in the population) influenced LDL-C by a magnitude below 10% of its standard deviation, which corresponds to <4 mg/dl. However, there is a well-described trade-off between variant frequency and effect size: while common variants tend to have small effects, rare variants can have very large ones, with some described to lower LDL-C by up to 50%. This spectrum, from rare variants with large effects to common variants with modest ones, makes the genetic evidence very informative, as we can study the consequences of LDL variation across its full range.
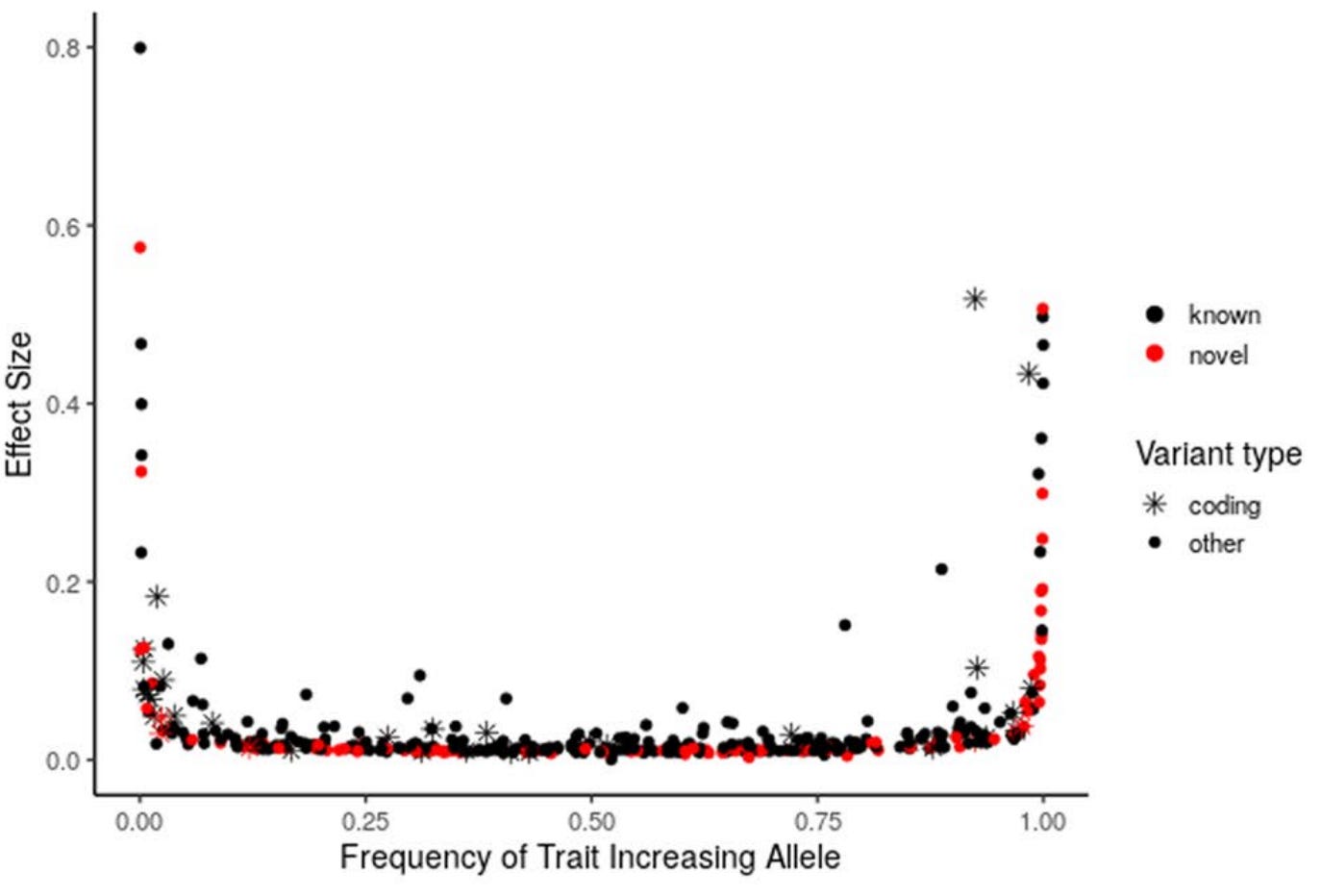
Natural experiment #1: rare variants with large LDL-increasing effects
The best-known piece of genetic evidence comes from studies of rare variants associated with large increases in LDL-C. Before GWASs, before we even had statins, we already knew about familial hypercholesterolemia (FH), a genetic condition associated with high cholesterol levels and a substantially higher risk of myocardial infarction earlier in life. FH is an autosomal dominant disorder. In the most common form, a loss-of-function mutation in the LDLR gene impairs the LDL receptor, so circulating LDL particles cannot be efficiently cleared from the bloodstream through the liver. Less commonly, the causes of FH are loss-of-function mutations in APOB (which codes the protein binding to the LDL receptor) or gain-of-function mutations in PCSK9 (a protein that normally degrades LDL receptors, and in its overactive form leaves even fewer receptors available).
Heterozygous carriers of one copy of FH mutations are relatively common in the general population. They represent roughly 1 in 200 to 1 in 300 people worldwide, making it one of the most common inherited disorders. Left untreated, it produces elevated LDL-C levels up to 500 mg/dl and leads to premature atherosclerosis and coronary artery disease (CAD).
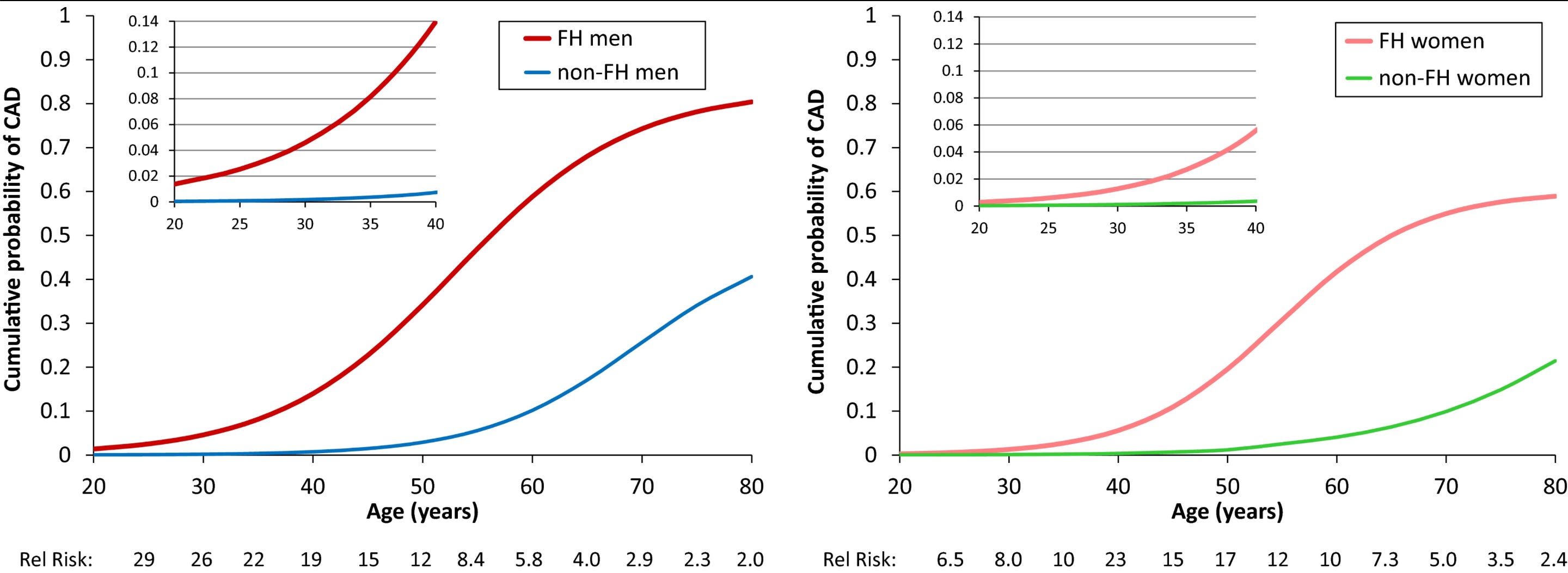
Beyond the elevated CAD risk, several observations among FH carriers are particularly suggestive of causality: (1) There is significant variation in LDL-C levels among carriers (between approximately 200 and 500 mg/dl) and this variation is associated with CAD risk and atherosclerosis burden in a dose-response pattern. (2) Despite different underlying molecular causes, the extent of atherosclerosis appears independent of which gene carries the mutation and dependent on the resulting LDL-C levels. (3) Within affected families, siblings who inherit the mutation compared to those who do not have a substantially higher risk of CAD proportional to their LDL-C levels, minimizing confounding from shared lifestyle. (4) Among individuals with FH, duration of exposure to high LDL-C, calculated through the simple index of cholesterol-years, is a better predictor of plaque burden than single measurements of LDL-C, highlighting the key concept of cumulative exposure.
Homozygous FH is far rarer: around 1 in 160,000–300,000. But the phenotype is extreme with untreated LDL-C often exceeding 500 mg/dl from birth. Historically, these patients developed atherosclerosis in childhood and suffered myocardial infarctions even before adolescence. It is hard to look at a child with blocked coronaries and an LDL-C of 700 mg/dl and argue that something else is responsible. The pioneers of cholesterol biology (and Nobel laureates) Goldstein and Brown discovered the LDL receptor largely by focusing their studies on patients with FH (this is a great 2009 review by Goldstein and Brown themselves on the chronicles of the discovery). It is worth pausing to acknowledge how much of modern cardiovascular prevention we owe to the study of these young patients.
Natural experiment #2: rare variants forcing large LDL reductions
The FH analyses show that large lifelong LDL-C elevation raises cardiovascular risk in a dose-dependent manner. But if LDL-C is causal, the mirror-image prediction would be that variants leading to lifelong LDL reduction should protect against atherosclerotic cardiovascular disease. We have such experiments, too.
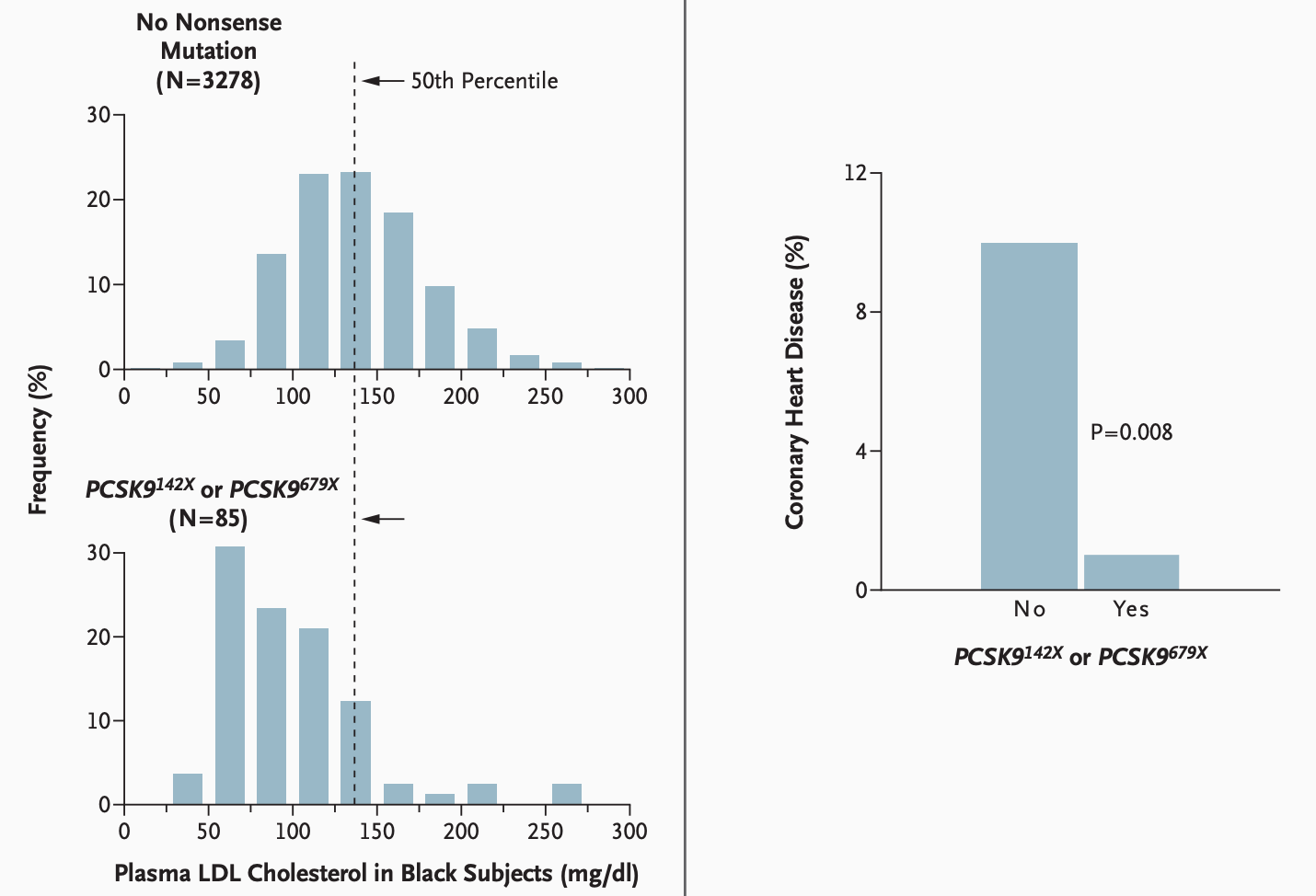
Although very rare in European ancestry populations, studies in individuals of African ancestry in the mid-2000s found that ca. 2% of the population carry a copy of a nonsense mutation in their PCSK9 gene. These mutations introduce premature stop codons resulting in a clear loss-of-function phenotype. As PCSK9 normally promotes LDL receptor degradation, its loss-of-function leads to more LDL receptors and as a result better clearance of circulating LDL (and other ApoB-containing) particles. Accordingly, and as one would expect, these individuals have lower levels of LDL-C. In the Atherosclerosis Risk in Communities (ARIC) study, the 85 carriers of the two loss-of-function mutations (all heterozygotes) had on average 37 mg/dl lower LDL-C than non-carriers. These individuals were less likely to develop CAD, with only 1 diagnosis over a follow-up period of 15 years (1.2%) compared to 9.7% among non-carriers, corresponding to an impressive hazard ratio of 0.11 (95% CI: 0.02 to 0.81).
Probably even more striking was the finding from another cohort, the Dallas Heart Study, of “an apparently healthy, fertile, normotensive, college-educated woman with normal liver and renal function tests who works as an aerobics instructor,” who had two different inactivating copies of PCSK9, making her a functional knockout for the gene. Her PCSK9 protein levels were not detectable and she had an LDL-C of only 14 mg/dl (for comparison, LDL-C in a newborn averages around 30 mg/dl at birth, rises to above 60 mg/dl by 2 months, and reaches approximately 100 mg/dl by adolescence)! Evidence that this represented a phenotype entirely compatible with normal life provided a critical boost to the development of pharmacotherapies targeting PCSK9. These drugs are now approved and used in clinical practice.
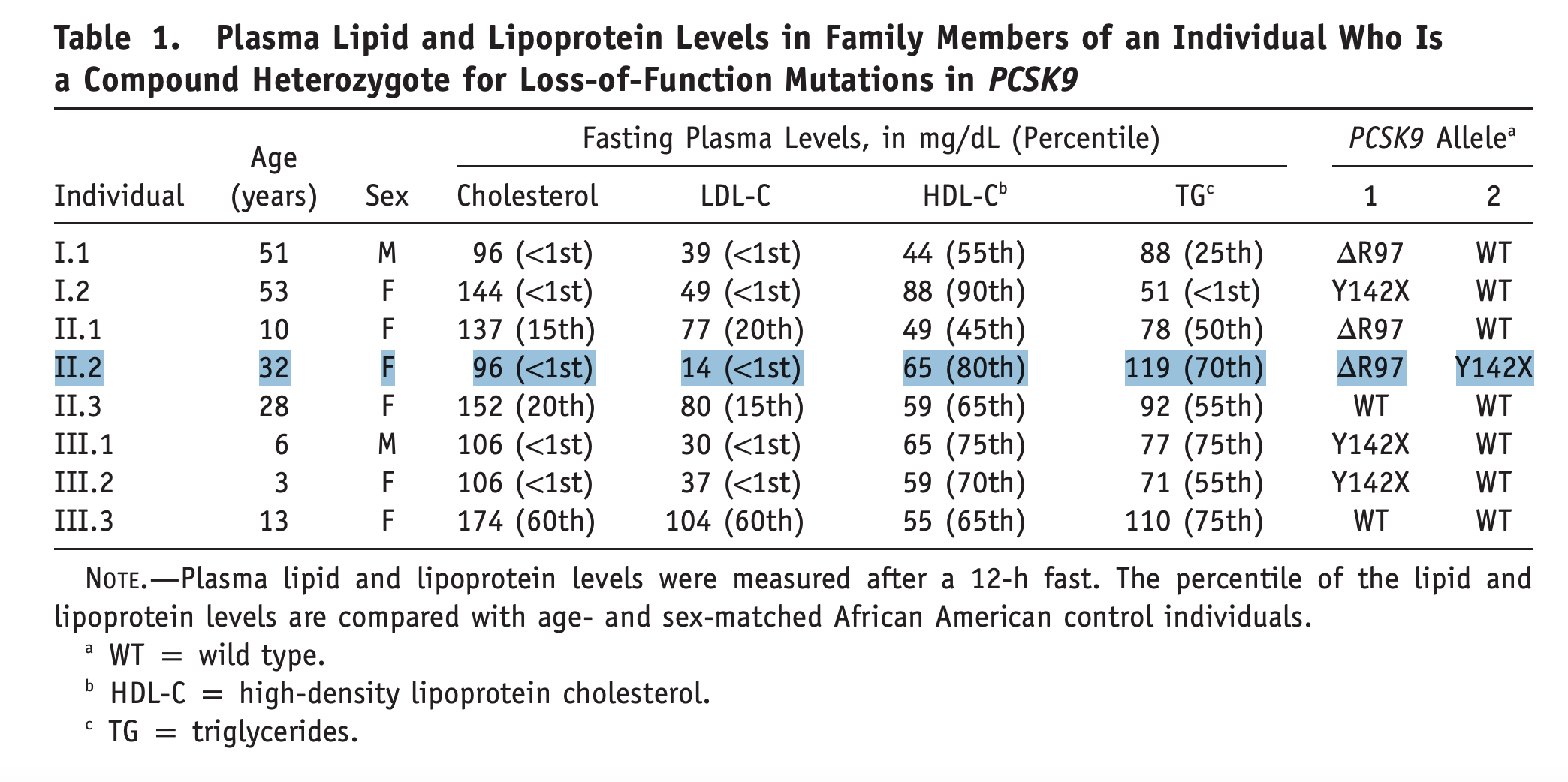
Later analyses confirmed these findings in additional cohorts, incorporating more PCSK9 rare variants with similar or less dramatic loss-of-function effects. Consistently, rare loss-of-function variants leading to lifelong lower LDL-C produced a proportional reduction in risk of CAD.
Meanwhile, the evidence extends beyond PCSK9 alone. Loss-of-function APOB variants impairing LDL receptor binding are associated with even larger reductions in LDL-C of approximately 55 mg/dl. In a Japanese cohort, these mutations were associated with an OR of 0.28 for CAD (95% CI, 0.12–0.64). Another example is NPC1L1, the target of ezetimibe involved in intestinal cholesterol absorption, where inactivating variants were associated with lower LDL-C and accordingly with lower risk of CAD (OR: 0.47, 95% CI: 0.25–0.87).
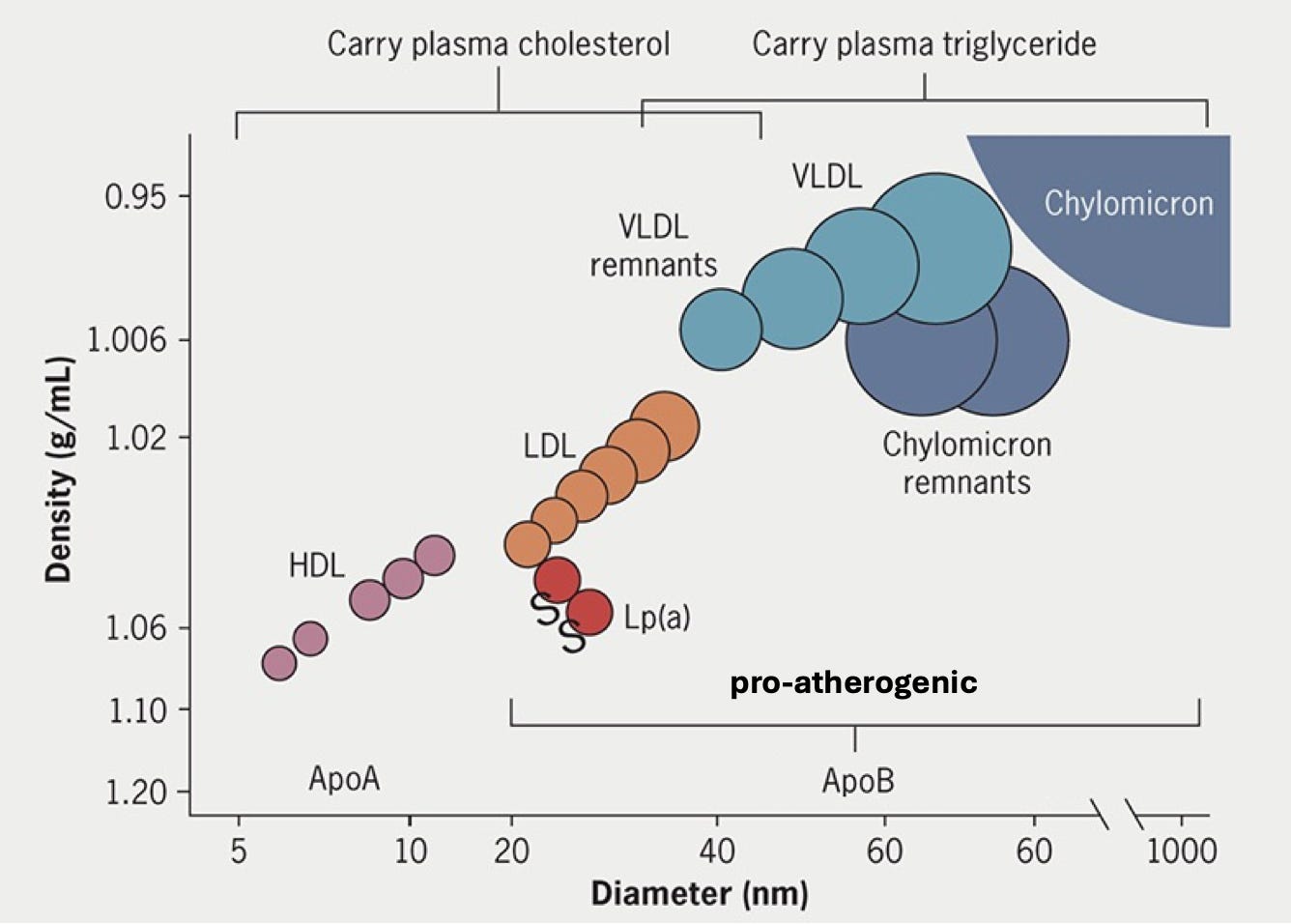
From LDL to ApoB-containing lipoproteins
Although LDL-C has been the focus so far, the biology of how lipoproteins cause atherosclerosis is more nuanced. All non-HDL lipoprotein particles carry one copy of the protein ApoB and are responsible for delivering cholesterol and fats to peripheral tissues. In the fasting state in metabolically healthy individuals, roughly 90% of these ApoB-containing particles are LDL, which predominantly carry cholesterol. The remaining fraction consists of larger triglyceride-rich particles, including VLDL, IDL, and chylomicron remnants, which are less abundant under normal conditions but rise substantially in obesity, type 2 diabetes, and metabolic syndrome.
Genetic findings from rare loss-of-function studies suggest these triglyceride-rich particles are also atherogenic. Loss-of-function variants in APOC3, ANGPTL3, and ANGPTL4, which encode inhibitors of lipoprotein lipase, the enzyme that breaks down triglycerides, lead to dramatically lower triglyceride levels with minimal changes in LDL-C. And yet carriers have meaningfully lower CAD risk. This provides the biological rationale for a wave of emerging therapies, with ANGPTL3 and APOC3 inhibitors already in phase 3 trials and ANGPTL4 inhibitors in phase 2.
Natural experiment #3: common variants in known lipoprotein metabolism pathways
Beyond rare variants with large effects, common variants in genes involved in lipoprotein metabolism are also associated with measurable effects on atherosclerotic cardiovascular disease risk, albeit with much smaller effects sizes.
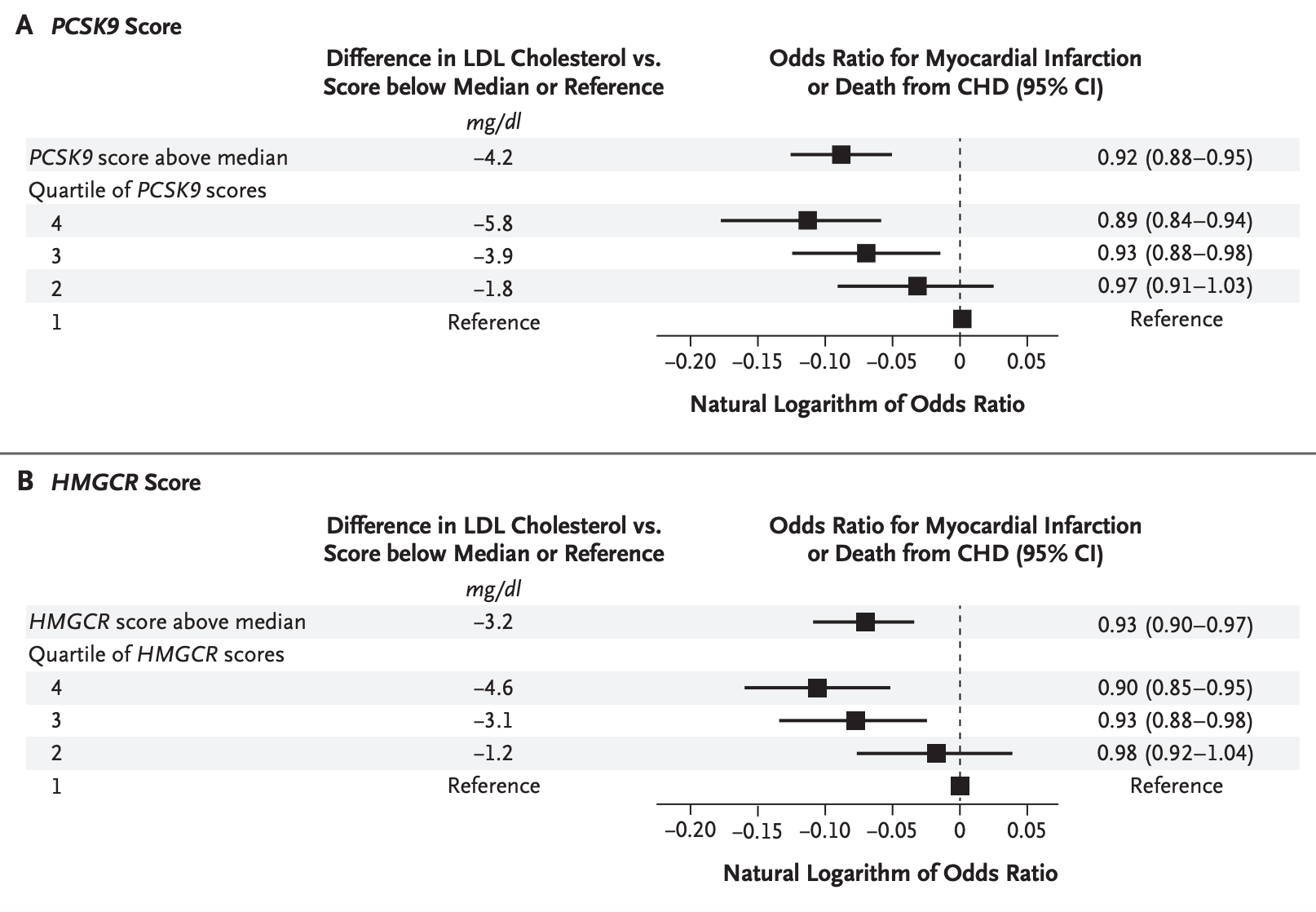
Consider HMGCR and PCSK9, the genes coding for the targets of statins and PCSK9 inhibitors, respectively. HMGCR codes for HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. Within the loci of these genes, several common variants are associated with lower LDL-C levels. It is unclear how each individual variant acts (presumably via modest effects on enzyme activity or gene expression), but several GWASs confirm consistent effects on LDL-C levels. Because these are common variants, individuals may carry several of them simultaneously. Weighting each variant by its effect on LDL-C, we can compute genetic scores for individuals in large population-based studies and test their association with CAD.
In such an experiment using data from around a decade ago, genetic variation linked with lower LDL-C in either gene (6 and 7 variants in the loci of HMGCR and PCSK9 respectively) was associated in a dose-response manner with lower odds of myocardial infarction. The effect sizes on CAD risk are expectedly smaller than what we saw for rare variants: individuals in the top versus bottom quartiles had an LDL-C difference of only 5.8 mg/dL for PCSK9 variants and 4.6 mg/dL for HMGCR variants, with corresponding ORs for myocardial infarction of 0.89 and 0.90. But the direction and dose-response pattern are fully consistent.
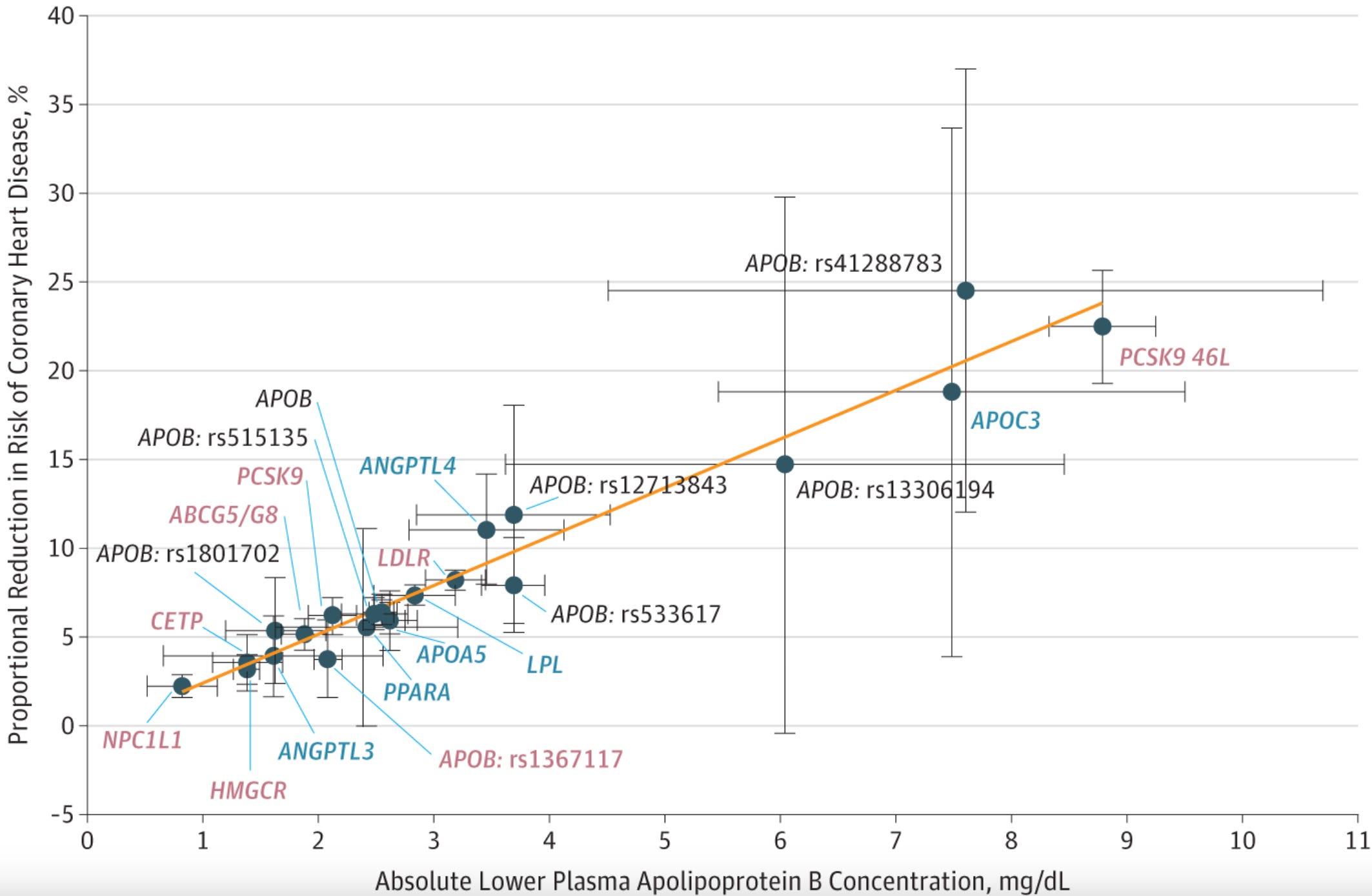
Similar results have since been observed for all genes known to be directly involved in LDL metabolism that are direct or indirect targets of existing or emerging therapeutics, including HMGCR, PCSK9, NPC1L1, CETP, ACLY, and LDLR. Notably, this pattern holds across both LDL-related and triglyceride-related genes (ANGPTL3, APOC3, PPARA, LPL), consistent with the hypothesis that both LDL and triglyceride-rich lipoprotein particles are atherogenic.
Instead of LDL-C or triglycerides, we can weight the effects of these variants by their influence on ApoB concentration rather than LDL-C or triglycerides, ApoB being the best proxy for the total number of atherogenic particles since each carries exactly one ApoB molecule. If we then plot these effects against CAD odds, they all fall on a strikingly straight line, corresponding to an OR of 0.78 per 10 mg/dL decrease in ApoB. This unifying picture holds across the full frequency spectrum: rare variants substantially raising ApoB-containing lipoproteins (as in FH), rare variants substantially lowering them (as with PCSK9 or APOC3 loss-of-function variants), and common variants influencing ApoB in either direction. In each case, the effect on CAD risk follows a clear dose-response pattern proportional to the effect on ApoB concentration, which reflects the number of circulating ApoB-containing particles
Natural experiment #4: common variants throughout the genome influencing LDL
This framework can be tested even more broadly beyond the genes already known to be involved in lipoprotein metabolism. GWASs have identified variants across hundreds of loci consistently associated with lipid measures, many acting through mechanisms we do not yet fully understand. This means we can run a broader genome-wide experiment: taking all variants linked to LDL-C or ApoB regardless of their mechanism of action and testing whether they influence CAD risk. Using data from the UK Biobank, this yields 252 common variants associated with LDL-C levels and 241 associated with ApoB concentration, spanning all 22 chromosomes and a highly diverse set of genes. When we plot the effects of these variants on lipids against their association with CAD odds, the picture is broadly consistent with the analyses restricted to known genes.
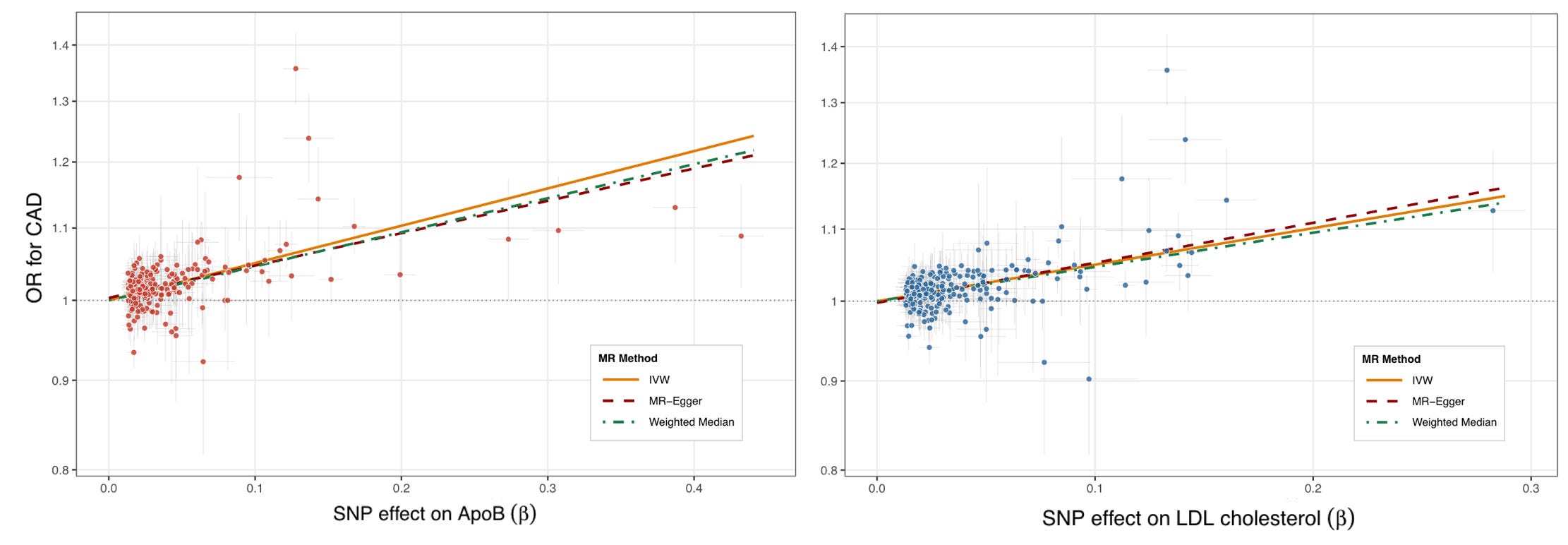
For comparison to the previous graph, the OR produced with the conventional inverse-variance weighted (IVW) approach is 1.64 per standard deviation increment in ApoB concentration. In the UK Biobank, the standard deviation of ApoB is 23.8 mg/dL, which scaled to a 10 mg/dL decrement gives an OR of 0.81 — very close to the estimate from variants in known pathways alone (0.78).
The two graphs are visibly messier than the previous one produced by Ference et al. This is captured in between-variant heterogeneity statistics, which are highly significant in both cases. This is to be expected as variants acting distantly on LDL-C or ApoB may also influence CAD through other mechanisms, contributing to pleiotropy. This risk is of course much lower when restricting to variants in genes directly involved in lipoprotein metabolism. Any pleiotropic effects can influence the results, if they consistently push the line in one or the other direction (directional pleiotropy). The association in this case remains consistent even when modeled with approaches that are less sensitive to pleiotropic variant effects (MR-Egger and the weighted median estimator).
A careful observer of the two graphs will notice that there are some SNPs that do not appear to follow the general pattern. This is especially evident in the upper left of the plots, where some variants appear to be associated with higher risk of CAD per unit increase in ApoB concentration that the overall trend would predict. When we look at which variants these are, the two most prominent ones map to the LPA locus and are known to also substantially raise Lp(a) levels. Lp(a) is an LDL-like ApoB-containing lipoprotein with an additional molecule of apolipoprotein(a) (coded by LPA), whose circulating levels are overwhelmingly genetically determined (heritability estimates of 70-90%), and consistently associated with higher CAD risk across multiple large-scale genetic studies.
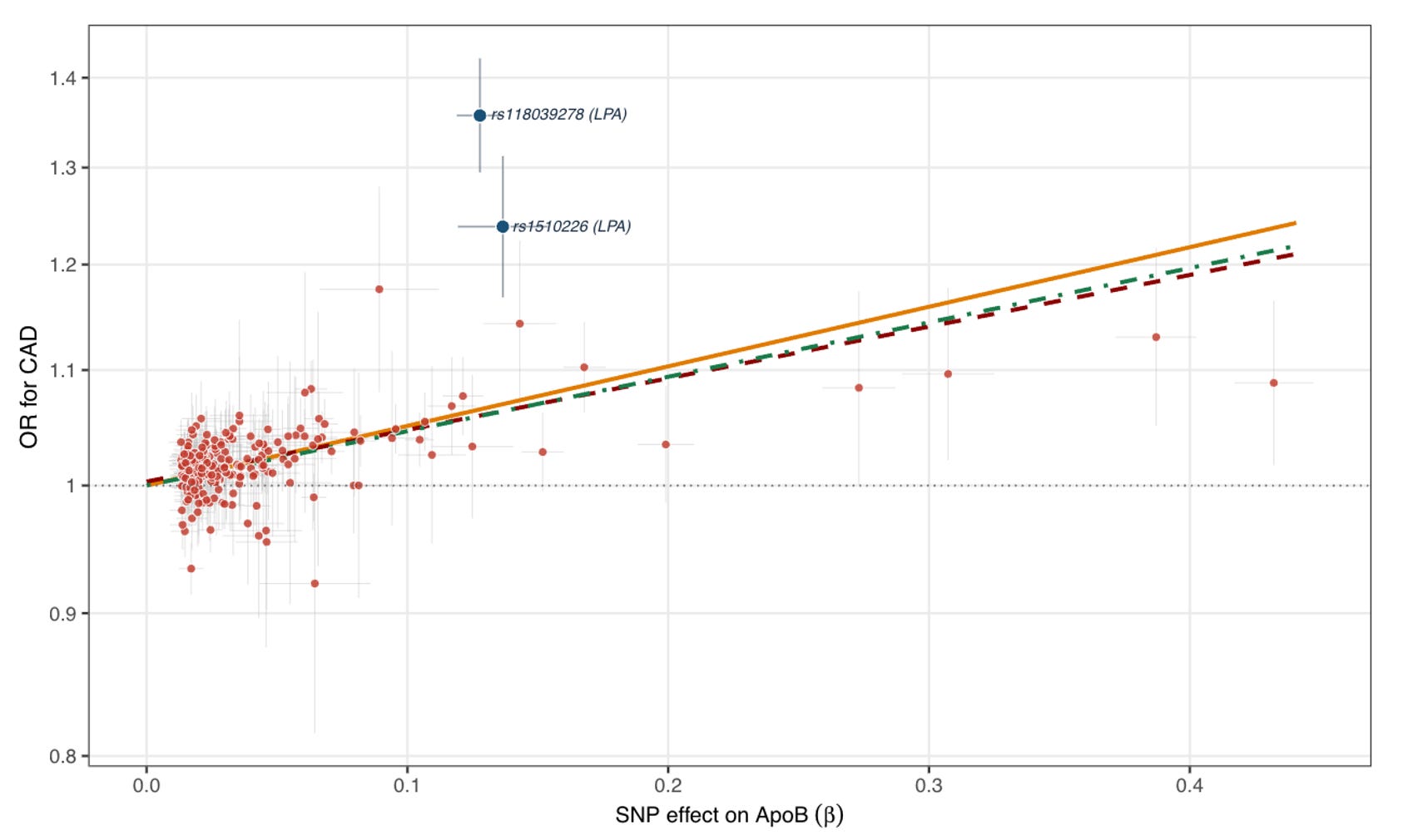
This deviation has been previously described and is an interesting side-story. In an analysis clustering variants by whether they preferentially raise Lp(a) versus LDL, the OR for CAD per unit ApoB was substantially larger for the Lp(a)-raising cluster, suggesting that Lp(a) particles are approximately 6-fold more atherogenic than traditional LDL particles. Lp(a) is not reduced by statins (if anything they increase it slightly) and only moderately by PCSK9 inhibitors, leaving it as a genetically driven risk factor without a targeted treatment to date. Drugs lowering Lp(a) by up to 90% are in advanced development, with phase 3 readouts expected as early as this year. Lp(a) probably deserves a separate discussion, as it is another example of how genetic evidence has shaped the current model of lipoprotein atherogenicity.
The breadth and specificity of the signal
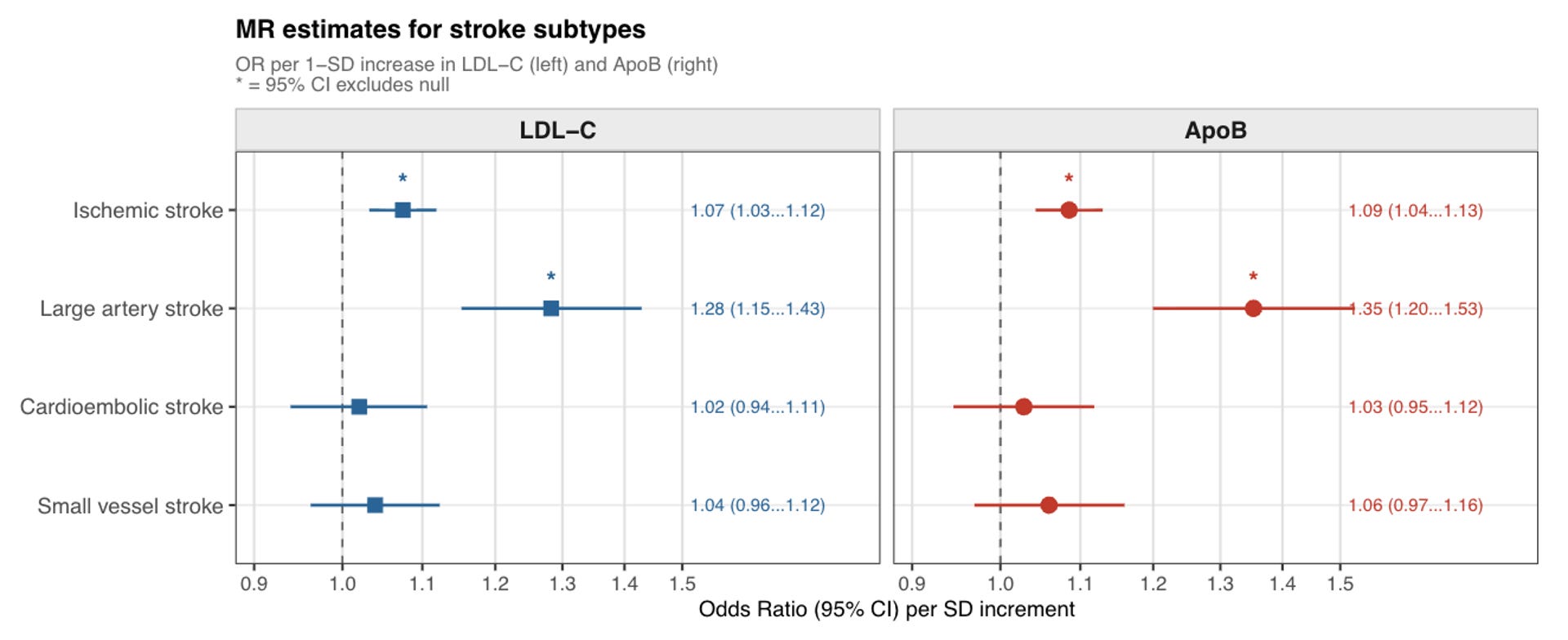
An important point to consider is that CAD is not the only clinical manifestation of atherosclerosis. Although most of the genetic evidence presented here was first established for CAD, more modern datasets have allowed exploration of a broader range of atherosclerotic phenotypes. Similar results have been observed for ischemic stroke, peripheral artery disease, carotid atherosclerosis, abdominal aortic aneurysm, and aortic stenosis, supporting the concept that ApoB-containing lipoproteins are drivers of atherosclerosis accumulation throughout the vasculature.
Ischemic stroke is a particularly instructive case, as it is a highly heterogeneous condition with multiple distinct underlying pathologies. This heterogeneity makes it a useful test of specificity: if the genetic signal truly reflects atherosclerosis, it should associate with atherosclerosis-related stroke subtypes but not with others driven by different mechanisms. This is precisely what the data show. Genetically proxied LDL-C and ApoB concentrations are associated with large artery stroke, which is attributed to atherosclerosis, but show weak or null associations with cardioembolic and small vessel (lacunar) stroke, which are driven by other pathologies. This subtype specificity strengthens the case for a causal atherosclerotic mechanism.
Genetic vs. trial estimates: cumulative burden of atherogenic particles
It is interesting to try to recapitulate the genetic findings with the results of randomized controlled trials of LDL-C-lowering treatments. Trials of statins and PCSK9 inhibitors consistently show approximately 20-25% relative risk reduction per mmol/l (38.7 mg/dl) lower LDL-C over five years of treatment. Mendelian randomization analyses, reflecting lifelong genetically lowered LDL-C, show risk reductions of approximately 50% for an equivalent absolute LDL-C reduction.
This indicates that the slope of the LDL-CAD relationship steepens with duration of exposure. Randomized trials intervene in middle age, after decades of high exposure to atherogenic lipoprotein particles that has led to accumulation of arterial damage, and run for a around three to five years. The genetic variants used in Mendelian randomization studies have been lowering LDL-C since conception, integrating over 50 or 60 years of exposure in the typical study participant. Consistent with trial data comparing efficacy across duration of treatment, this has contributed to the now widely accepted hypothesis of cumulative exposure to atherogenic lipoproteins as the principal driver of atherosclerosis accumulation and cardiovascular risk.
If this is true, it has considerable implications for clinical practice. Rather than treating only people with advanced atherosclerosis at very high risk of cardiovascular events, the cumulative exposure model argues for keeping atherogenic lipoprotein particle burden as low as possible for as long as possible. The genetic evidence, by showing that lifelong lower LDL-C produces substantially larger risk reductions than short-term pharmacological lowering later in life, provides perhaps the strongest argument for earlier and more sustained intervention.

Predicting trial results from genetic data
A final point worth highlighting is that the field of lipidology has set the standard for using human genetic data not only to establish causal biology but to directly inform therapeutic target prioritization and drug development. Human genetics are often credited with transforming drug development, yet most therapeutic areas cannot point to any approved or emerging drugs that were the direct result of genetic findings. Cardiovascular risk reduction is a striking exception. PCSK9 inhibitors are approved and widely used. Lp(a)-targeting therapies are approaching phase 3 endpoint readouts. Treatments targeting ANGPTL3 and APOC3 are in phase 3 studies, and ANGPTL4 inhibitors are in phase 2.
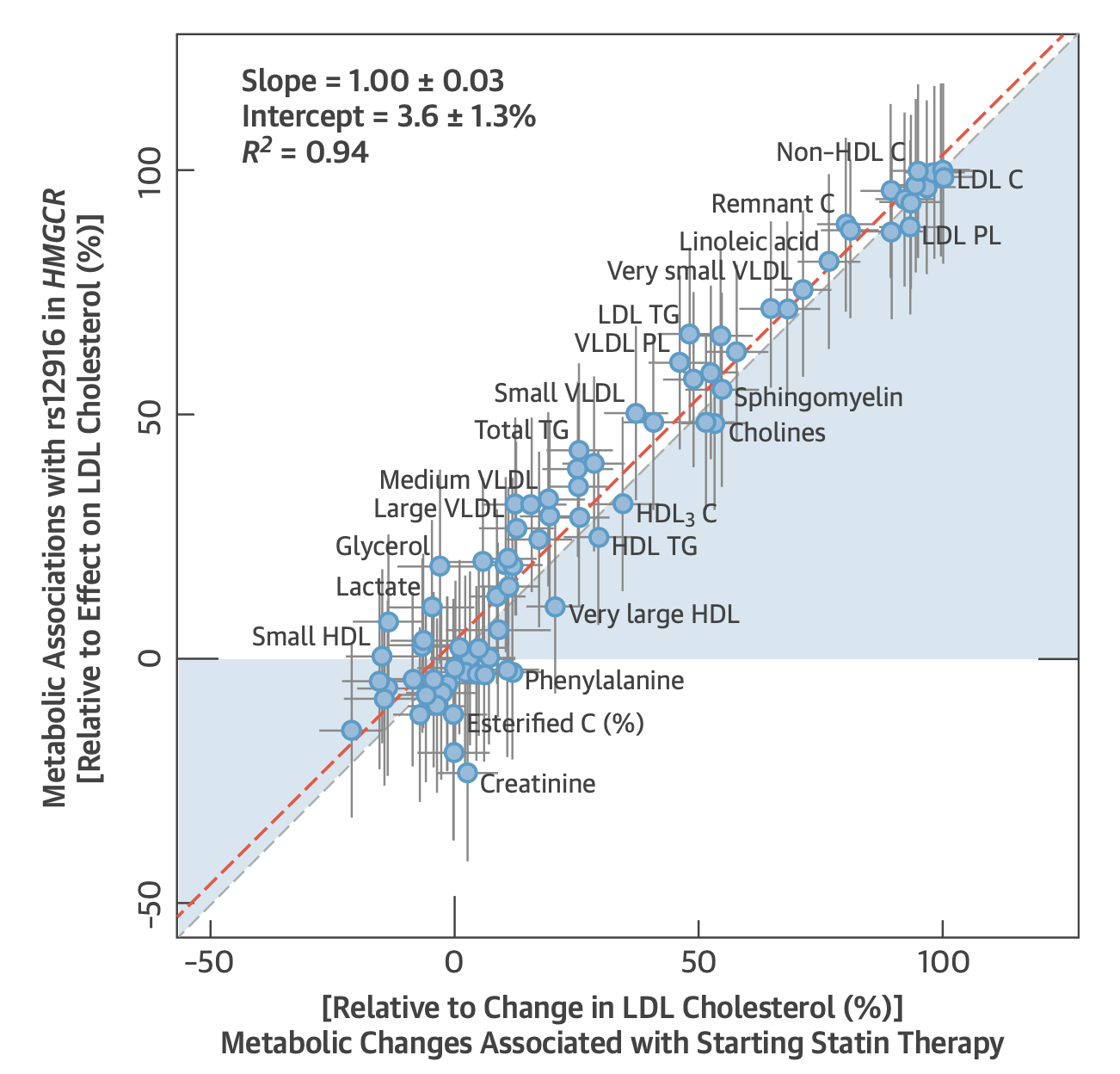
Beyond new targets, the genetic framework has also successfully recapitulated the effects of existing drugs. Common variants in HMGCR mirror the effect of statins on cardiovascular disease risk, genetically validating their target long after they entered the market. In fact, genetic variation in HMGCR almost perfectly mimics the metabolic effects of statins across multiple metabolomic and lipidomic measurements (graph below) and even predicts diabetes as a side-effect. Perhaps most compellingly, common and rare variation in NPC1L1 leading to lower LDL-C was associated with lower CAD risk, predicting the results of the IMPROVE-IT trial shortly before it was published. The same logic now extends to reductions of triglyceride-rich lipoproteins and Lp(a), where genetic evidence is strong. The ongoing trials will tell us whether the framework's predictive record holds.
Concluding thoughts
What the genetic data collectively suggest is that atherosclerosis accumulation and cardiovascular risk are driven by the duration of exposure to atherogenic ApoB-containing lipoproteins. This appears to operate along three axes: LDL particles, which represent the largest pool and the primary target of existing therapies; triglyceride-rich lipoproteins, increasingly recognized as causal contributors particularly in the context of obesity and metabolic disease; and Lp(a) particles, which are highly atherogenic but present in meaningful quantities only in a genetically defined subset of individuals. Addressing all three long before irreversible arterial damage has accumulated would represent the most rational strategy to maximally reduce atherosclerotic cardiovascular risk.
Put simply, human genetic evidence does not support any meaningful form of a cholesterol debate. Every independent line of genetic evidence converges on the same highly consistent model: more LDL (or ApoB) means more atherosclerosis, and less means less.
Crucially, this holds for almost any variant in the human genome that influences ApoB, regardless of whether it has other metabolic effects. HMGCR variants that lower ApoB also raise glucose and diabetes risk; yet, they still reduce cardiovascular risk. Many PCSK9 and LDLR variants lower LDL-C and ApoB exclusively, with no effect on triglycerides or HDL; yet, they still reduce cardiovascular risk. Most variants have no relationship with obesity or insulin resistance whatsoever; yet, they still reduce cardiovascular risk.
Of course, this does not mean that there are no important questions open. Why do some variants deviate from the general dose-response pattern, and what does this tell us about heterogeneity in particle atherogenicity? How do other cardiovascular risk factors such as hypertension, smoking, diabetes, and obesity interact with lipoprotein particle burden to modify individual risk? Can these risk factors initiate and propagate atherosclerosis even in individuals with very low numbers of atherogenic lipoproteins? What is the optimal intervention point, and how should we track atherosclerosis accumulation across the vasculature? Are there any side-effects associated with very large reductions of LDL-C or ApoB sustained over very long periods of time that clinical trials might be too short or underpowered to detect? For example, genome-wide selected variants linked to lower LDL-C levels in both European and Chinese ancestry populations, HMGCR variation, and genetic variation linked to larger on-statin LDL-C reduction, have all been associated with higher intracerebral hemorrhage risk. This signal seems to be largely dismissed in the cardiology community in view of several trials reporting no such risk with intensive LDL-C-lowering and only weak signals in earlier statin trials. The genetic data suggest it may deserve more careful attention than it currently receives, especially in high-risk populations or if we consider intensive LDL-C-lowering starting in young asymptomatic individuals as a preventive strategy.
More broadly, the LDL case has set the standard for how human genetic data can be used to establish causal biology and prioritize therapeutic targets, shaping the entire field of genetics-informed drug development. I am often asked why human genetics seems more widely implemented in cardiovascular drug development than in other fields. Part of the answer lies in historical tradition (don’t forget that the field of lipoprotein biology was built on the study of a rare genetic disorder), but foresight has also played a role: cardiovascular genetics researchers made the deliberate effort to translate complex genetic findings into clinically relevant language. For example, the direct comparisons of effect sizes between genetic studies and randomized trials, which strict methodologists might reasonably critique for conflating lifelong genetic exposures with short-term pharmacological ones, served as an effective bridge between the genetics and clinical communities. In many other therapeutic areas, human genetics remains a methodologically dense niche, poorly understood by the clinical researchers who ultimately run the trials that validate targets and hypotheses.