
Factor XI inhibition: from human genetic signal to the clinic
Asundexian moving toward approval for secondary stroke prevention

Recently, the phase 3 OCEANIC-STROKE trial reported that the activated factor XI inhibitor asundexian reduced recurrent ischemic stroke and major cardiovascular events without increasing intracranial or major bleeding among patients with non-cardioembolic ischemic stroke or high-risk transient ischemic attack. These results are important for several reasons. Clinically, they introduce a new pillar for secondary stroke prevention beyond antiplatelet therapy, statins, and vascular risk factor control in a very large and highly vulnerable patient population. Scientifically, they represent a major milestone for factor XI therapeutics that found an entry point to the clinic after decades of research promising a safer anticoagulant strategy.
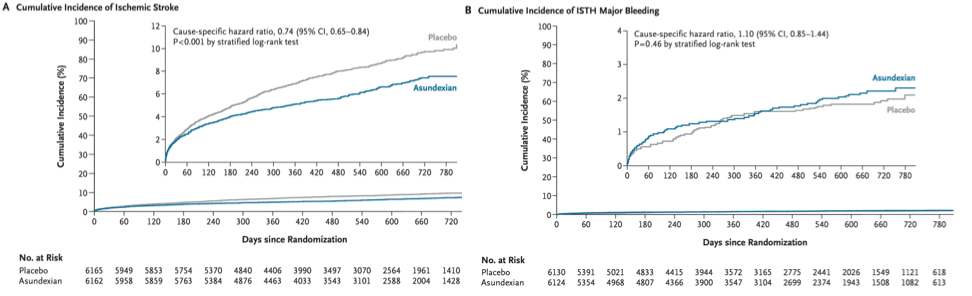
But the results are also fascinating from the perspective of human genetics. Factor XI inhibition represents a very clear example of a therapeutic success story that was strongly supported by genetic evidence long before clinical trial readouts. Studies of both rare factor XI deficiency and common F11 variation influencing factor XI levels or activity consistently pointed toward fewer thrombotic events with modest bleeding risk.
Rare F11 variation: inherited factor XI deficiency
Factor XI is a coagulation factor within the intrinsic pathway of the coagulation cascade. Activated by factor XII, factor XI amplifies downstream thrombin and fibrin formation, contributing to thrombosis and hemostasis.

Mutations in the gene coding for factor XI (F11 on chromosome 4), have long been recognized in the context of hemophilia C, a rare bleeding disorder typically inherited in an autosomal recessive pattern and affecting approximately 1 in 100,000 people worldwide. As opposed to hemophilia A or B that are caused by deficiencies of factors VIII and IX, respectively, hemophilia C is associated with a substantially milder bleeding phenotype. In fact, many individuals with factor XI deficiency remain asymptomatic throughout life, with the condition often identified only after unexpected bleeding during surgery or incidentally on routine coagulation testing in individuals without a prior bleeding history.
This unusually mild bleeding phenotype has important therapeutic implications. While the coagulation cascade has long been targeted pharmacologically, a central limitation of anticoagulation has been the trade-off between antithrombotic efficacy and bleeding risk. Thus, factor XI emerged as an attractive target. However, because factor XI deficiency has only mild effects on bleeding, it could be argued that factor XI may be partially dispensable in coagulation, raising the concern that its inhibition could have limited efficacy in preventing thrombotic events.
With such a low frequency in the general population, it is difficult to assess whether factor XI deficiency is associated with thrombotic risk. However, as is often the case for rare variants, certain historically isolated populations can show substantially higher frequencies. One well-known example for factor XI deficiency is the Ashkenazi Jewish population, where prevalence has been reported to be as high as 1 in 450, with heterozygosity up to 9%!
Utilizing this elevated prevalence, a previous study leveraged an Israeli population-based electronic record system to examine whether factor XI activity is associated with thrombotic outcomes. Factor XI activity had been measured in 10,193 adults between 2002 and 2014. Activity of 50% or lower was associated with reduced cardiovascular events (stroke, transient ischemic attack, or myocardial infarction), with hazard ratios of 0.51 (95% CI: 0.30–0.87) for activity of 30–50% and 0.57 (95% CI: 0.35–0.92) for activity below 30%. The effect was even more pronounced for venous thromboembolism, with only 3 events (0.2%) in individuals with reduced factor XI activity compared with 94 (1.1%) among those with activity above 50%, corresponding to an adjusted hazard ratio of 0.26 (95% CI: 0.08–0.84).
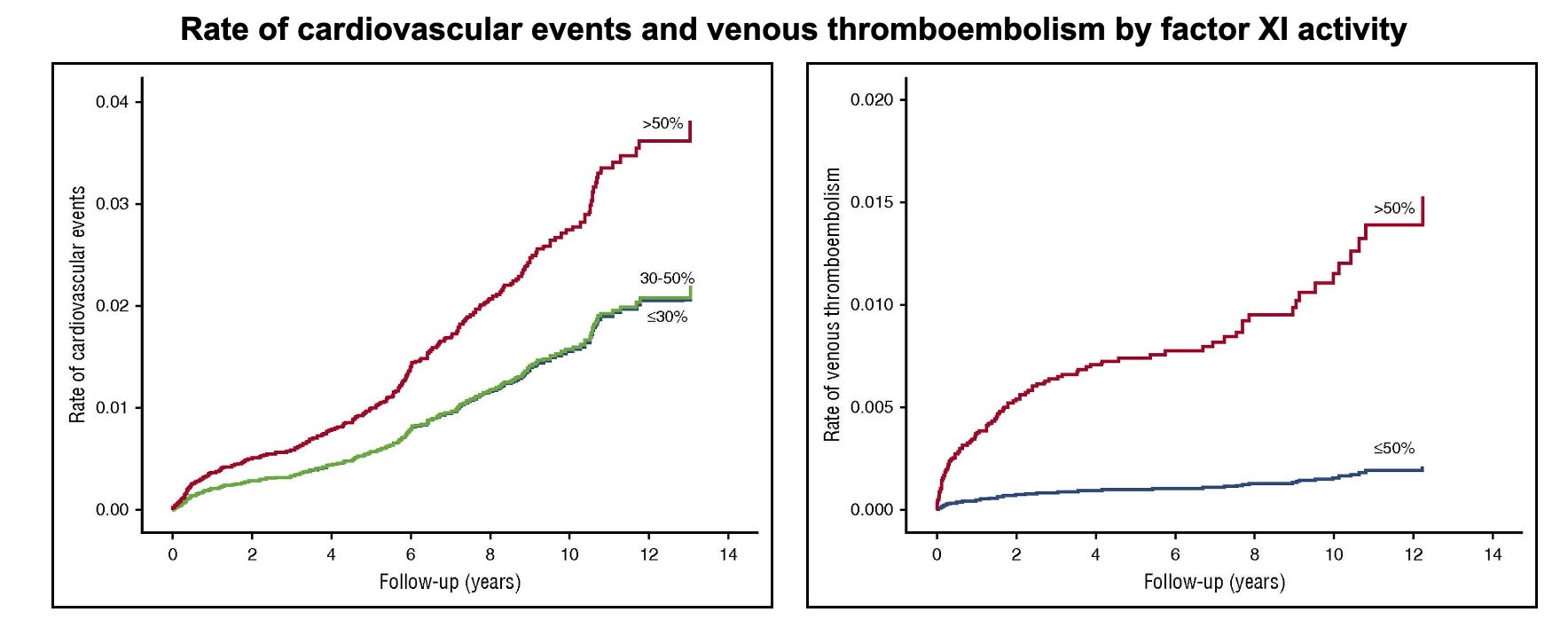
This finding largely agreed with earlier, smaller studies (also in Ashkenazi Jewish cohorts), which reported reduced incidence of ischemic stroke and venous thromboembolism among individuals with severe factor XI deficiency compared to general population estimates. Interestingly, no such association was observed for myocardial infarction.
Nowadays, large-scale whole-exome sequencing datasets allow direct assessment of whether coding variation in F11 is associated with thrombotic and bleeding outcomes in larger populations. Using the UK Biobank, we can identify individuals carrying predicted loss-of-function or damaging F11 variants and test their association with clinical endpoints. In the European ancestry subset of the UK Biobank (approximately 450,000 individuals), 0.7% carry one copy of a rare, predicted deleterious F11 coding variant (including protein-truncating and damaging missense or splice-site variants; REVEL ≥ 0.25; minor allele frequency ≤ 0.001; source).
When tested against clinical outcomes, most associations do not reach genome-wide significance, largely reflecting limited power given that variant carriers among cases are typically fewer than 100 for most endpoints. However, restricting attention to prespecified thrombotic and bleeding phenotypes reveals some interesting patterns.
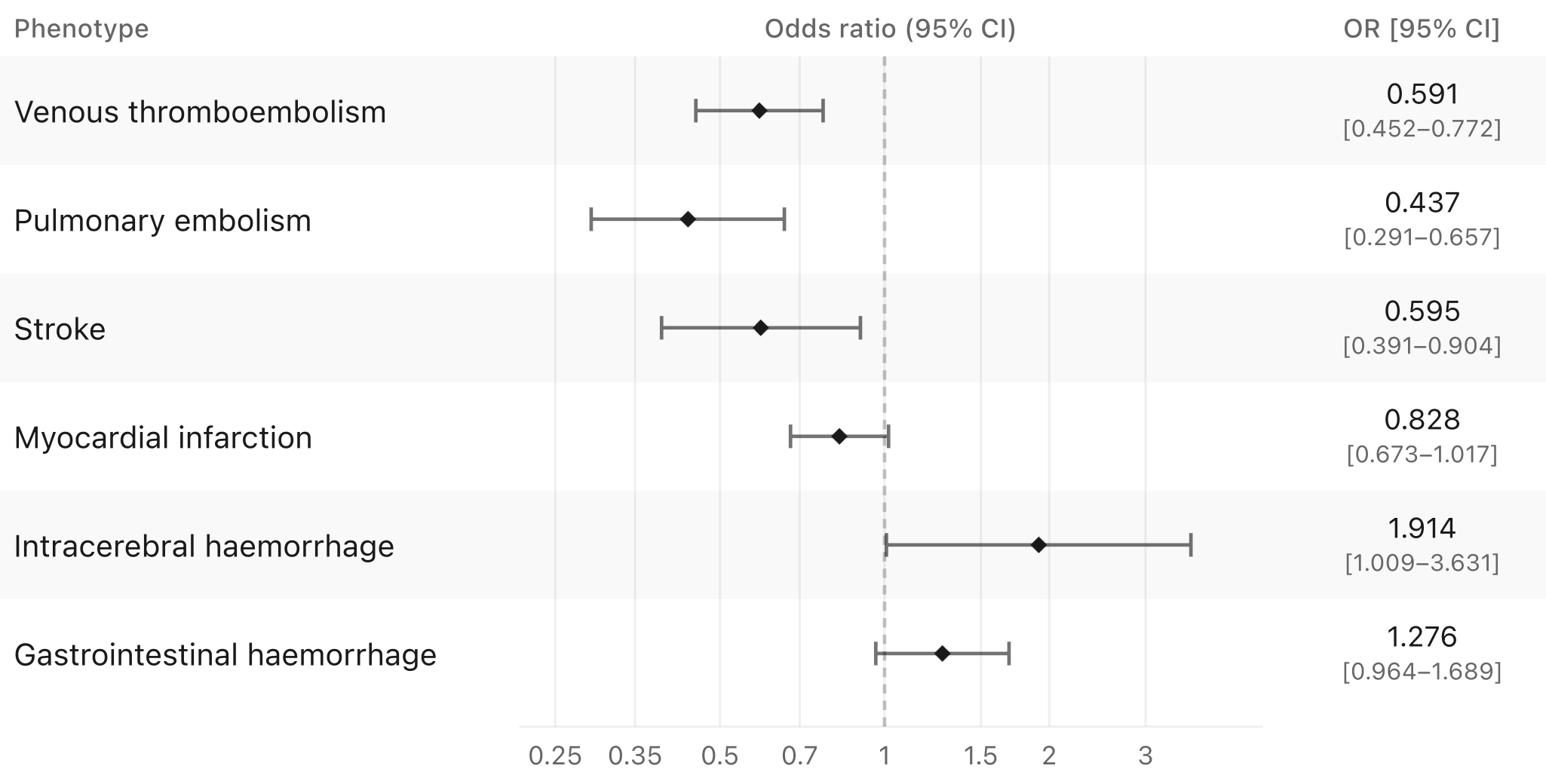
There is evidence of lower lifetime risk of venous thromboembolism, pulmonary embolism, and ischemic stroke. The association with myocardial infarction is weaker. As one would expect, there is some suggestive evidence for higher risk of clinically relevant bleeding outcomes, particularly intracerebral hemorrhage and gastrointestinal bleeding, but this does not reach significance and is not as large as one would expect for a population with a bleeding disorder.
Common variation in F11 and risk of bleeding and thrombotic endpoints
Rare variants provide powerful biological insight, but are often underpowered for robust association testing with disease endpoints. In contrast, large-scale genome-wide association studies (GWAS) enable the study of common variants with smaller effects on factor XI levels or activity. As we have GWAS data for both molecular traits and clinical outcomes, we can link variants directly influencing factor XI levels and activity with thrombotic and bleeding endpoints in a Mendelian randomization framework.
In the first such analysis, we utilized two independent variants in the F11 locus that were associated with FXI levels in a GWAS of 16,169 European individuals. rs4253417 is an intron variant in the F11 gene with the C allele (frequency ca. 30% among Europeans) raising factor XI levels by ca. 7.5%. The second variant rs62350309 is less common (ca. 5% in Europeans) and the A allele influenced factor XI levels by ca. 3.5%. Pooling the two variants and scaling their effects to units increase in factor XI levels showed an association with higher ischemic stroke risk, but also no convincing evidence for myocardial infarction. The association with intracerebral hemorrhage was not significant, but largely underpowered.
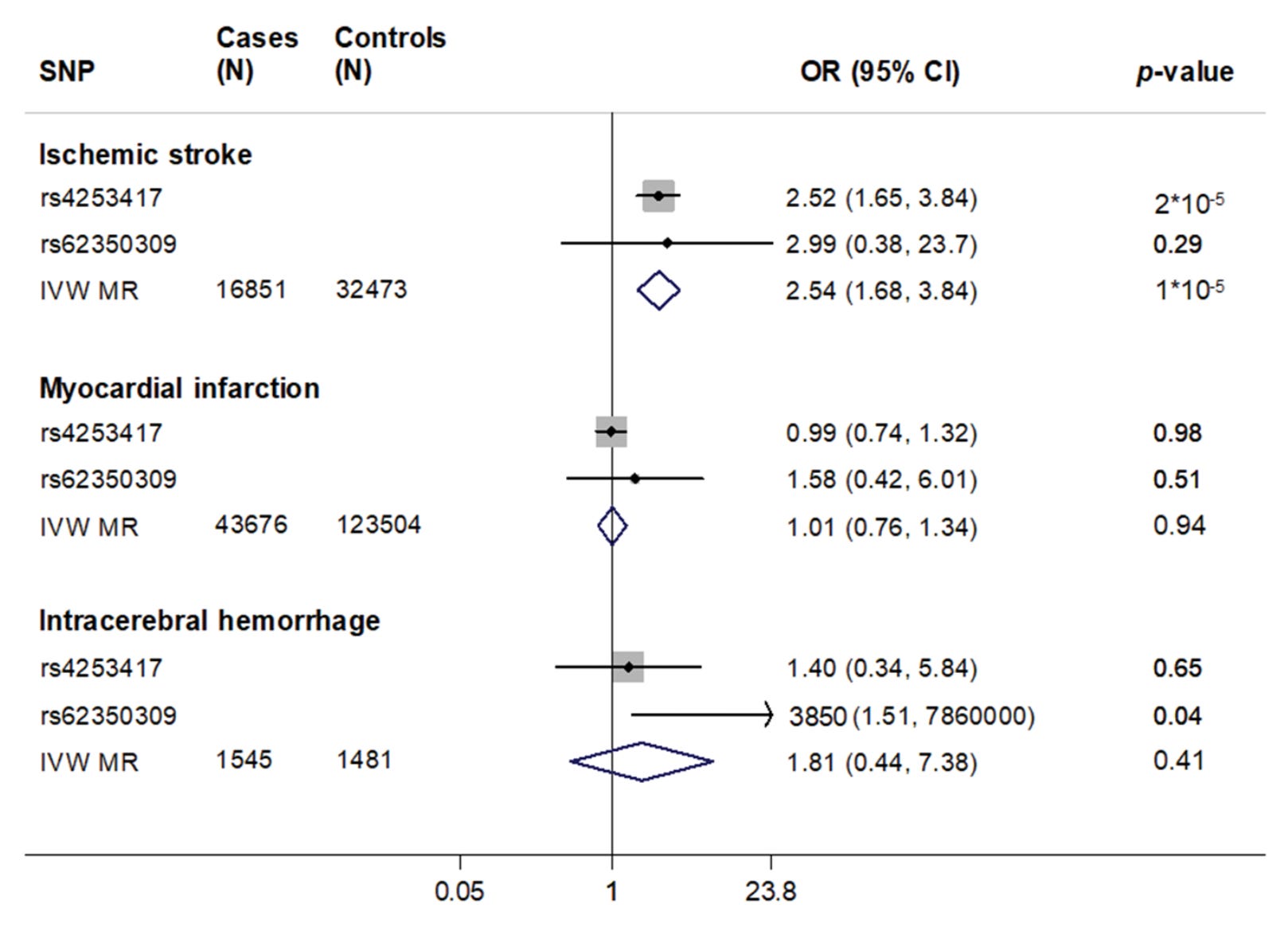
Since this analysis was published, variants in the F11 locus, including rs4253417, have reached genome-wide significance level (p<5E-8) in larger GWASs of both ischemic stroke (e.g. here) and venous thromboembolism (e.g. here). Subsequent Mendelian randomization analyses in larger datasets have largely replicated these findings.
Repeating this analysis with contemporary datasets allows the use of substantially larger numbers of cis-acting variants associated with factor XI levels. Using proteomic measurements from UK Biobank and deCODE, and selecting genome-wide significant variants associated with FXI levels measured by either Olink or SomaScan, we identify 38 and 28 variants at modest linkage disequilibrium (r2 < 0.1), respectively. This enables more granular Mendelian randomization analyses with substantially improved statistical power.
Testing these variants against ischemic stroke, myocardial infarction, venous thromboembolism, intracerebral hemorrhage, and gastrointestinal bleeding produces a pattern that increasingly resembles the rare variant analyses. Genetically predicted lower factor XI levels are associated with reduced risk of ischemic stroke and venous thromboembolism, with substantially stronger effects for the latter. In contrast, there is little evidence for myocardial infarction, alongside modest increases in risk of intracerebral hemorrhage and gastrointestinal bleeding.
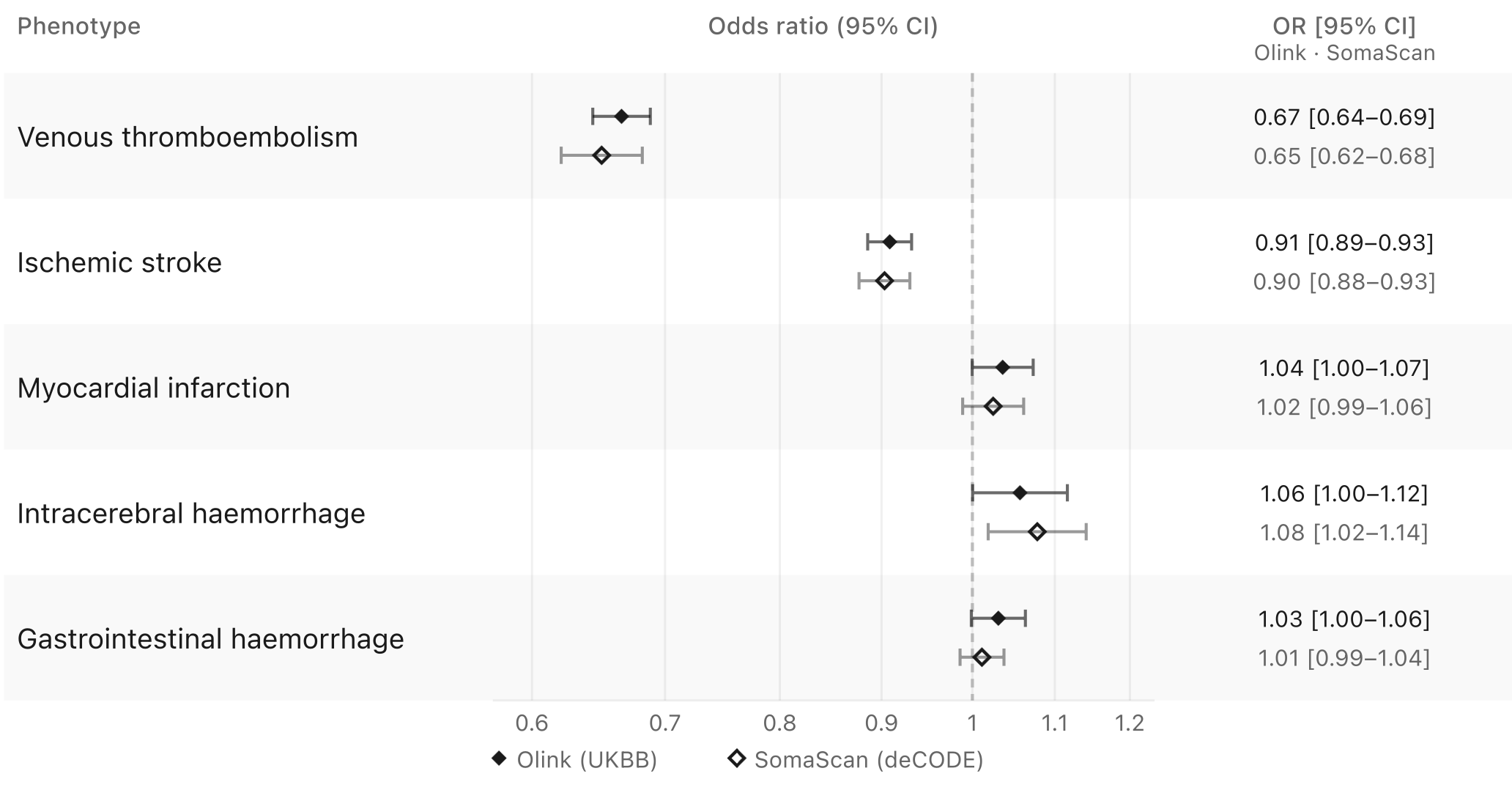
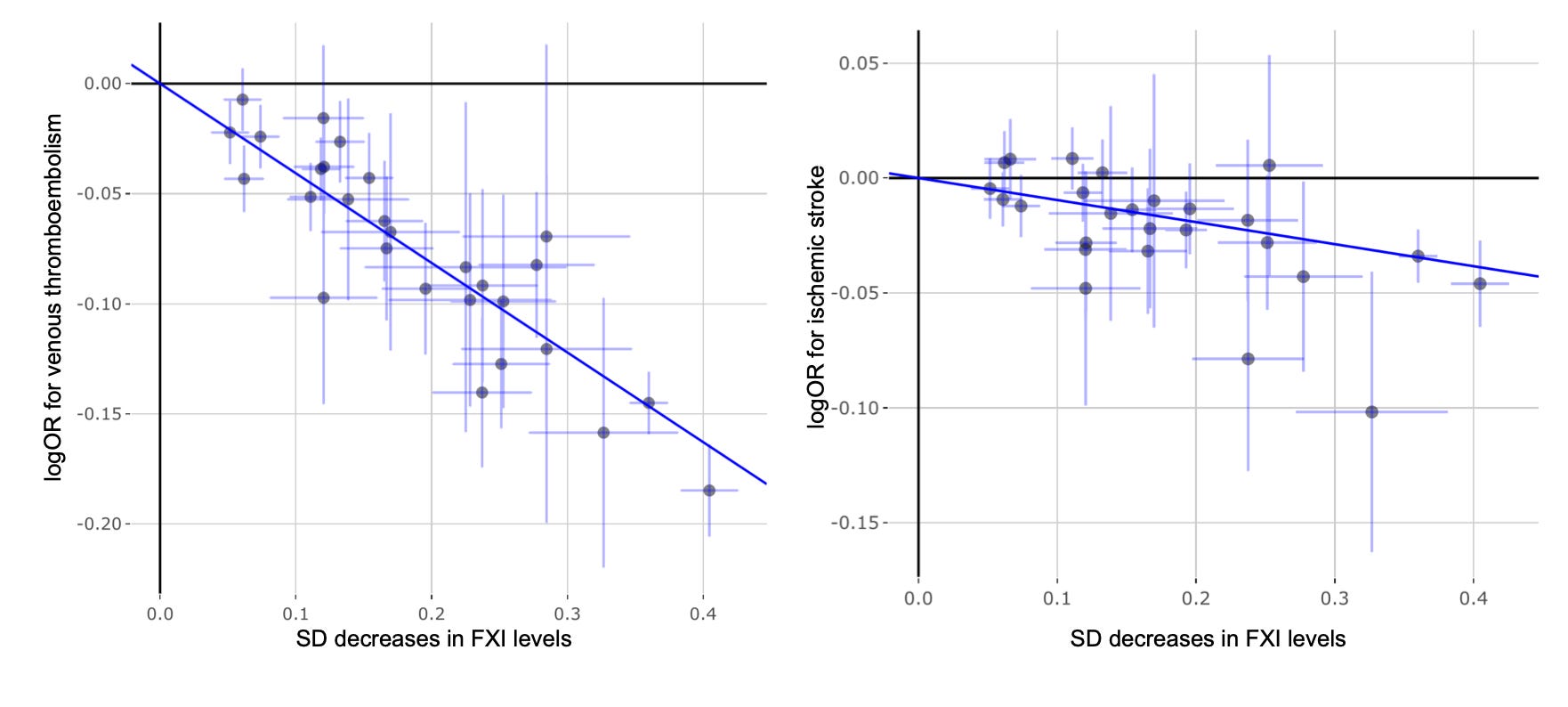
If we examine individual variants, the signal for venous thromboembolism becomes remarkably clear. Across 28 F11 variants available in the venous thromboembolism dataset, there is an almost perfectly log-linear relationship between their effects on Olink-quantified factor XI levels and lifetime venous thromboembolism risk (p = 1.2E-126). This degree of concordance is exceptionally rare in human genetics studies. Typically, some variants emerge as outliers because they influence disease risk through alternative pathways, but there is little evidence of that here. The corresponding analysis for ischemic stroke is also strongly significant (p = 1.6E-13), although the pattern is messier. This is not unexpected. Unlike venous thromboembolism, ischemic stroke is a highly heterogeneous phenotype encompassing multiple biological mechanisms, including atherosclerotic plaque rupture, cerebral small vessel disease, and atrial fibrillation-related cardioembolism, which may not all be equally influenced by factor XI biology. Nevertheless, the association remains consistent even when we model it with alternative approaches that account for heterogeneous effects of individual variants.
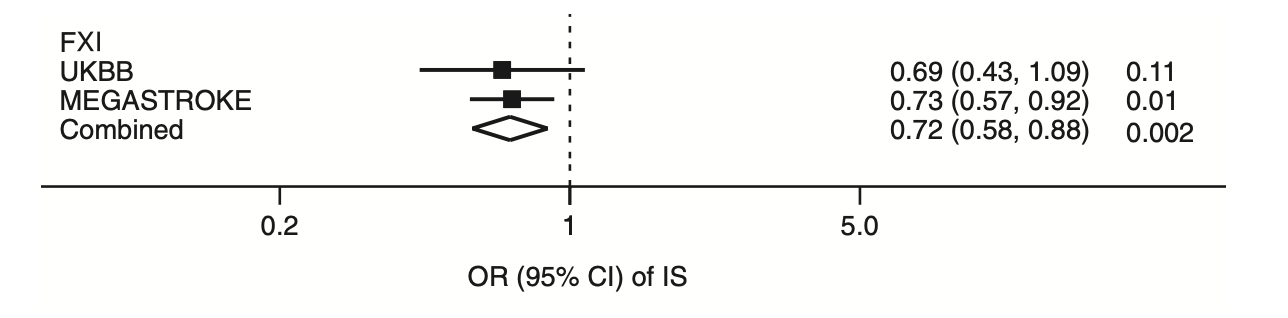
Variation within the F11 locus can influence factor XI biology through mechanisms beyond circulating factor XI levels alone. Some variants may directly affect factor XI activity independently of plasma levels, for example through missense changes altering protein function. Although large-scale genomic studies using specialized factor XI activity assays are not yet available, genomic data for activated partial thromboplastin time (aPTT) from Biobank Japan provide an indirect measure of intrinsic pathway activity. Notably, the variant associated with reduced aPTT in this dataset (rs56810541) was also associated with lower ischemic stroke risk.
An especially interesting aspect of the story, particularly when considered alongside the clinical trial results discussed below, is ischemic stroke subtype specificity. Ischemic stroke is a highly heterogeneous disease arising from distinct vascular pathologies. Genetic evidence supports associations across most major stroke subtypes, although the signals appear strongest for cardioembolic and cryptogenic stroke, as shown below.
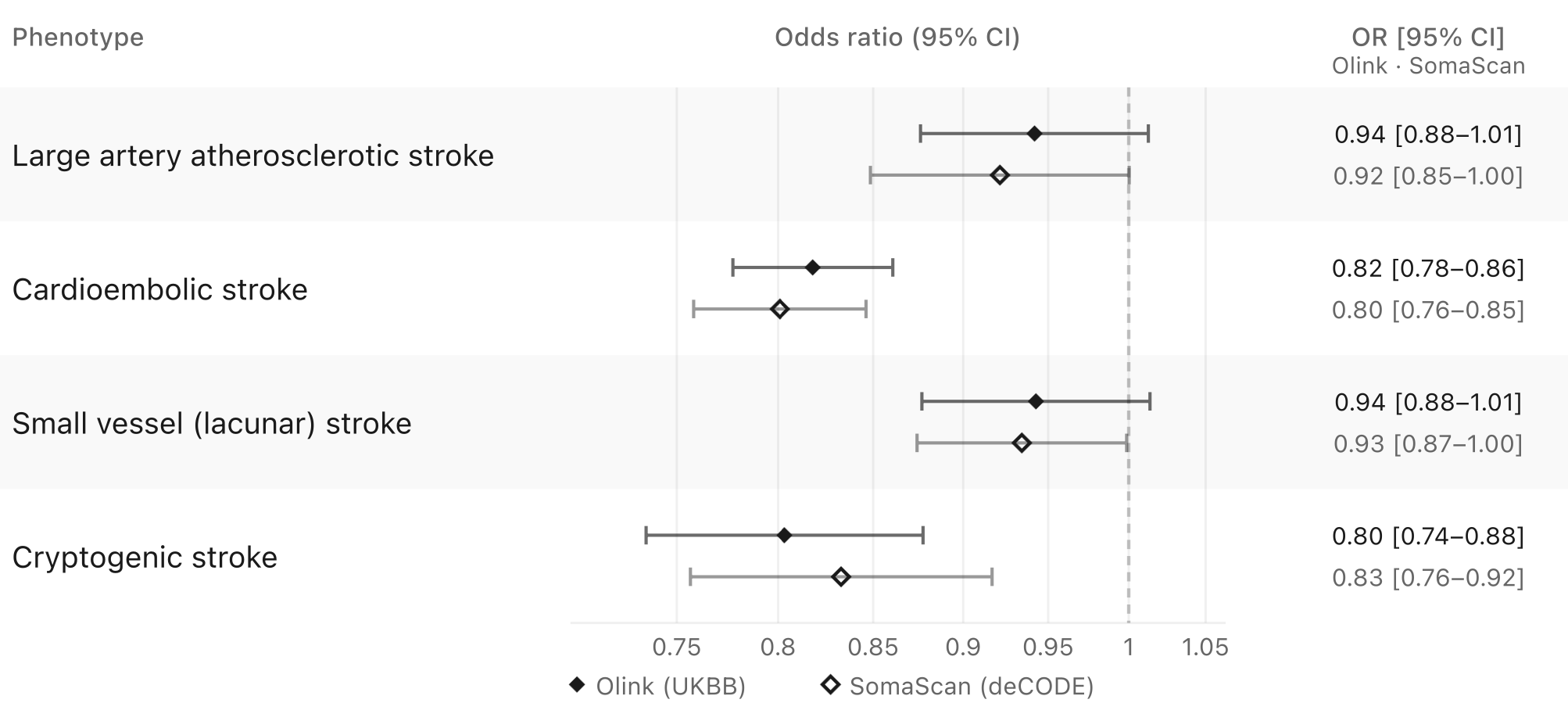
Importantly, these analyses should be interpreted cautiously. Compared with datasets for overall ischemic stroke, GWASs of stroke subtypes remain substantially underpowered, with approximately tenfold fewer cases. In addition, stroke subtyping requires detailed clinical phenotyping that is not consistently available across large biobanks and genetic consortia. Unlike clinical trials, which apply harmonized diagnostic protocols across sites, genetic studies are more susceptible to subtype misclassification.
Recapitulation with clinical trials
How do these genetic findings compare with clinical trials targeting factor XI? Several FXI-directed therapeutics are currently in late-stage development, including the oral FXIa inhibitors asundexian (Bayer) and milvexian (BMS, J&J), as well as abelacimab (Novartis), an antibody targeting factor XI. Among these, asundexian became the first to show success in a phase 3 trial. Based on positive results of the OCEANIC-STROKE trial, Bayer was granted priority review by the FDA and we should expect approval relatively soon.
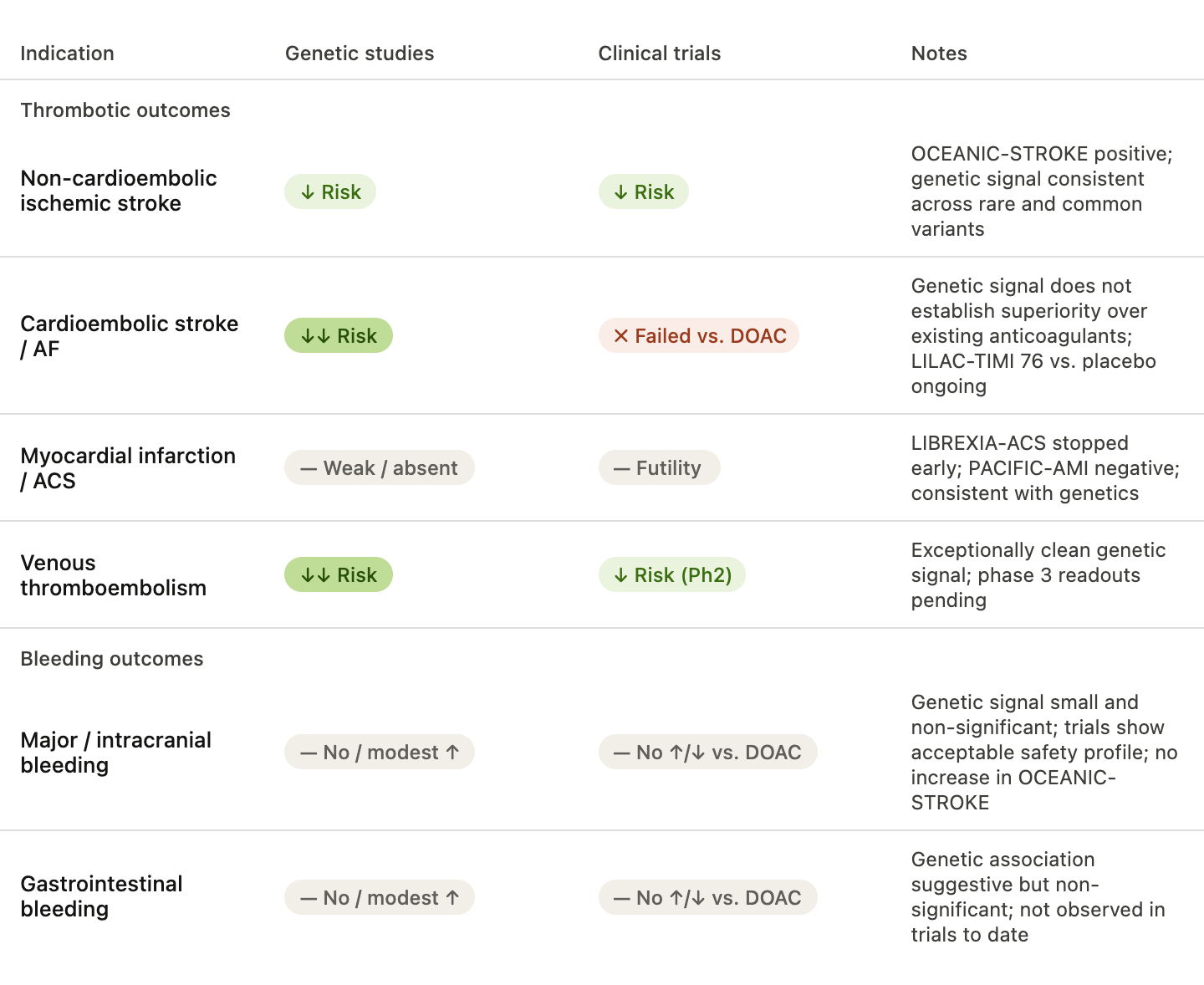
In OCEANIC-STROKE, asundexian reduced recurrent ischemic stroke and major cardiovascular events among patients with non-cardioembolic ischemic stroke or high-risk transient ischemic attack, without increasing major or intracranial bleeding risk. Interestingly, subgroup analyses suggested benefit across all major stroke subtypes, including large artery, small vessel, and cryptogenic stroke. This appears somewhat broader than the genetic data, where the strongest signals were observed for cardioembolic and cryptogenic stroke.
Cardioembolic stroke presents a particularly interesting case. Genetically, it appears to be the stroke subtype most strongly linked to factor XI biology. Yet both asundexian and abelacimab failed in the OCEANIC-AF and AZALEA–TIMI 71 studies of atrial fibrillation-related stroke prevention, with both trials stopped prematurely because of excess ischemic stroke in the investigational arm despite lower bleeding rates. Importantly, these were not placebo-controlled comparisons. Patients with atrial fibrillation already receive highly efficacious anticoagulants targeting factor Xa or thrombin, which inhibit coagulation further downstream. Human genetics can support the causal relevance of a pathway and predict effects relative to baseline biology, but it does not establish superiority over other already effective therapeutic strategies. An interesting case is the LILAC-TIMI 76 trial that tests abelacimab versus placebo in patients with atrial fibrillation who have been deemed unsuitable for oral anticoagulation due to excess bleeding risk. The study is ongoing and will provide a placebo comparison that is closer to genetic study, but also a potential solution to a group of patients that are at an otherwise high risk of ischemic stroke.
The broader trial landscape has been remarkably concordant with the genetic data. Consistent with the weak or absent genetic associations for myocardial infarction, the phase 3 study LIBREXIA-ACS testing milvexian in patients presenting with acute coronary syndromes to reduce risk of major adverse cardiovascular events vs. placebo was stopped prematurely due to futility. The phase 2 PACIFIC-AMI for asundexian also did not point to any reduction of major adverse cardiovascular events among patients with an acute myocardial infarction (although it was underpowered for it). Interestingly, there was no increase in bleeding risk although milvexian was added on top of double antiplatelet therapy. In contrast, two phase 2 thromboprophylaxis studies for milvexian and abelacimab after knee surgery have shown promising results for both venous thromboembolism and bleeding risk vs. enoxaparin that is standard therapy, matching the exceptionally strong genetic evidence for this endpoint. Phase 3 readouts for cancer-related venous thromboembolism are pending.
Regarding adverse bleeding effects, the results of all trials to date were consistent with an acceptable safety profile, and substantially lower bleeding risk when compared to other oral anticoagulants. This is largely concordant with the genetic data suggesting, if any, only small lifetime increases in bleeding risk for patients with genetically downregulated factor XI activity. Importantly, this was also the case for intracranial bleeding in OCEANIC-STROKE. As patients with ischemic stroke are at risk of bleeding in the ischemic brain tissue, this is clinically very important.
Concluding thoughts
With a successful phase 3 trial and a pending approval that is likely to change clinical practice for a very large group of patients, factor XI inhibition joins a select group of therapeutic successes supported by human genetic data that were collected years before clinical trial readouts. For cardiovascular outcomes, it now stands as evidence that the paradigm works beyond LDL-lowering biology.
That said, it would be reductionist to claim that genetics fully predicted the complex story of clinical development spanning a decade of trials. For example, the failures in atrial fibrillation, despite a strong genetic signal for cardioembolic stroke, were a consequence of comparing against already highly effective treatments, a challenge that genetics cannot anticipate. The futility in acute coronary syndromes, on the other hand, was entirely consistent with the weak genetic signal for myocardial infarction, and could have tempered enthusiasm for that indication earlier.
This makes factor XI an instructive case study, not just as a validation of human genetics, but as a reminder that human genetic evidence is not always directly translatable to clinical success. Clinical practice is complicated and integrating genetic findings to inform and accelerate drug development can be very challenging with each case having its own nuance. This is why analyses that pool all development efforts together to prove the value of human genetic support for drug development remain just an interesting thought exercise for pitch decks. Factor XI shows both why genetics matters and why it is not the whole story.